






Kinetics of Photocatalysis Reactions Studied by Transient Absorption Spectroscopy
- Photogenerated charge carriers in a semiconductor can be studied by Transient Absorption (TA) spectroscopy.
- The LP980 Transient Absorption Spectrometer provides spectral and time-resolved information on TiO2 photocatalysis.
- TA data show that photogenerated holes in TiO2 decay in the microsecond scale with complex kinetics.
- The photocatalytic oxidation of chloride, taking place in the millisecond range, is easily probed using the LP980 spectrometer.
Introduction
Photocatalysis is the rate increase of a chemical reaction by light, often in the presence of a catalyst that starts the reaction upon irradiation. Photocatalysts are typically semiconducting metal oxides such as ZnO, Fe2O3 or TiO2 which are employed as particles in solution. When absorbing light, these materials are able to generate electrons and holes which go on to react with chemical species on their surface.
The photocatalysis process is illustrated in Figure 1. If a photon of greater energy than the bandgap of the semiconductor is absorbed by the photocatalyst, it excites an electron (e–) from the valence band into the conduction band. This process generates a hole (h+) or positive charge in the valence band. These charge carriers are mobile, albeit short lived, and can evolve through multiple pathways. The electron-hole pair can recombine emitting energy after a short time, but the charge carriers can also be trapped at specific sites in the material. If they do not recombine, electrons and holes can react with species on the surface of the photocatalyst. Valence band electrons can combine with electron acceptors in solution, and conduction band holes may react with electron donors. This is often the first step in a series of charge-transfer reactions in solution that lead to a desired product.
Figure 1: Charge carrier generation in a photocatalyst particle. Conduction band electrons react with electron acceptors (A) and valence band holes react with electron donors (D).
Although photocatalysis has been known since the early 20th century, its first major practical application was not found until 1972 when Honda and Fujishima reported the photocatalytic splitting of water in the presence of titanium dioxide, TiO2.1 Since then TiO2 has established itself as a widely used photocatalyst. Despite its large bandgap requiring UV irradiation to be activated, it has some desirable properties such as high stability, high reactivity and low price.
The fate of the photogenerated carriers in a photocatalyst depends on the material, particle size and the presence of defects. Research on TiO2 and other photocatalysts focuses on increasing their efficiency by varying these parameters. Studying the kinetics of charge carrier transfer and recombination is a basic step in the design of photocatalytic materials: the longer the lifetime of the electron-hole pair, the better their chance of taking part in reactions. Some recombination events proceed via emission of light and can be followed by photoluminescence spectroscopy, however there is a large proportion of nonradiative recombination (i.e. non-emitting) events. For this reason, transient absorption (TA) spectroscopy is often used to follow the evolution of photogenerated charge carriers in TiO2. Charge carrier lifetimes can range from ps to ms, so they should be studied with a TA instrument covering a broad range of timescales. In addition, it is possible to follow the kinetics of photocatalytic reactions by monitoring the transient absorption from reaction intermediates.
In this application note, an Edinburgh Instruments LP980 Transient Absorption Spectrometer (Figure 2) is used to probe the dynamics of photogenerated charge carriers in a suspension of TiO2 particles. A hole-accepting species such as chloride is shown to be oxidised by activated TiO2, and the time-dependent absorbance of the dichloride radical is used to elucidate its reaction kinetics.

Figure 2: Edinburgh Instruments LP980 Transient Absorption Spectrometer.
Experimental
A titanium dioxide suspension was prepared by diluting a stock solution (Plasmachem, anatase, 10% wt) in deionised water to a concentration of 0.5% wt. Chloride-containing samples were prepared by adding HCl to this suspension to a concentration of 0.5 N. The suspensions were placed in quartz cuvettes and measured in an LP980 Transient Absorption Spectrometer using a standard TA sample holder (L-F01). The instrument was equipped with a 150 W pulsed Xe probe lamp, PMT-900 detector and ICCD camera. A frequency-tripled Nd:YAG laser producing 355 nm pulses at 10 Hz was used as the pump. The pulses were 6 ns long and were attenuated to an energy of 1.5 mJ/pulse.
TA curves were produced by averaging multiple transients in the L900 software. Fluorescence background induced by the pump laser pulse was characterised by blocking the probe beam and acquiring “pump-only” data. Similarly, fluctuations in the intensity of the probe beam were accounted for by recording “probe-only” data. “Pump-only” and “probe-only” data were automatically acquired and subtracted by the L900 software. The decay kinetics were fitted to multi-exponential components using the tail fitting algorithm included in L900.
Results
Transient absorption of the TiO2-only solution is caused by electrons and holes in the material produced by the pump beam. Free charge carriers recombine or are trapped in a few picoseconds, but trapped species can be detected by nanosecond TA.2 Trapped holes at the surface of TiO2 are most likely localised at oxygen sites forming O•- radicals which account for most of the TA spectrum, whereas trapped electrons are present in Ti3+ sites.2,3 Their spectra overlap forming a broad absorbance band in the visible: trapped electrons absorb at visible/red wavelengths, whereas trapped holes have a wide spectral signature from 300 nm to 800 nm. The ICCD detector in the LP980 spectrometer enables the acquisition of the full TA spectrum in a single shot, as shown in Figure 3. The result agrees with previous reports on the transient absorption spectrum of TiO2.4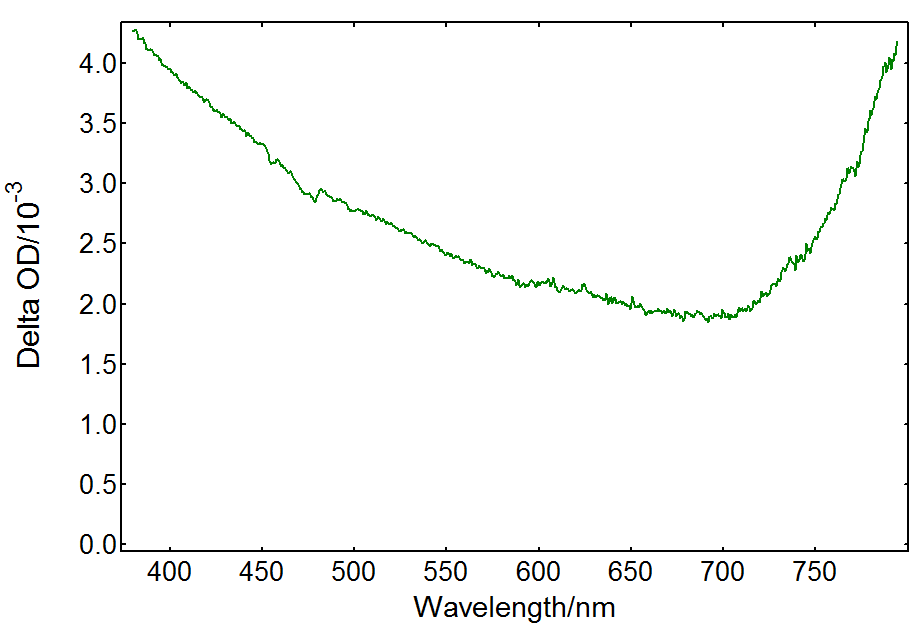
Figure 3: Transient absorption spectrum from a TiO2 suspension acquired in an LP980 spectrometer with an ICCD detector. Measurement conditions: λpump = 355 nm, monochromator bandwidth = 0.50 nm, ICCD delay = 100 ns, integration time = 1 µs, 100 averages.
The PMT detector on the LP980 spectrometer allows acquiring detailed time-dependent data at a particular wavelength. Figure 4 shows the TA decay from a TiO2 suspension at 390 nm, a transition assigned to trapped holes. The decay kinetics were fitted to an exponential model with n components:
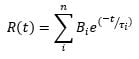
In the above equation t is the time after the pump pulse, τi is the lifetime of component i and Bi is a preexponential factor. The data in Figure 4 fits to a three-component decay with lifetimes of
21 ns, 188 ns and 3.29 µs. This complex kinetics suggests multiple pathways for the decay of photogenerated holes. The fact that the lifetime of trapped holes extends into the microsecond scale indicates that the material is suitable for photocatalysis reactions.
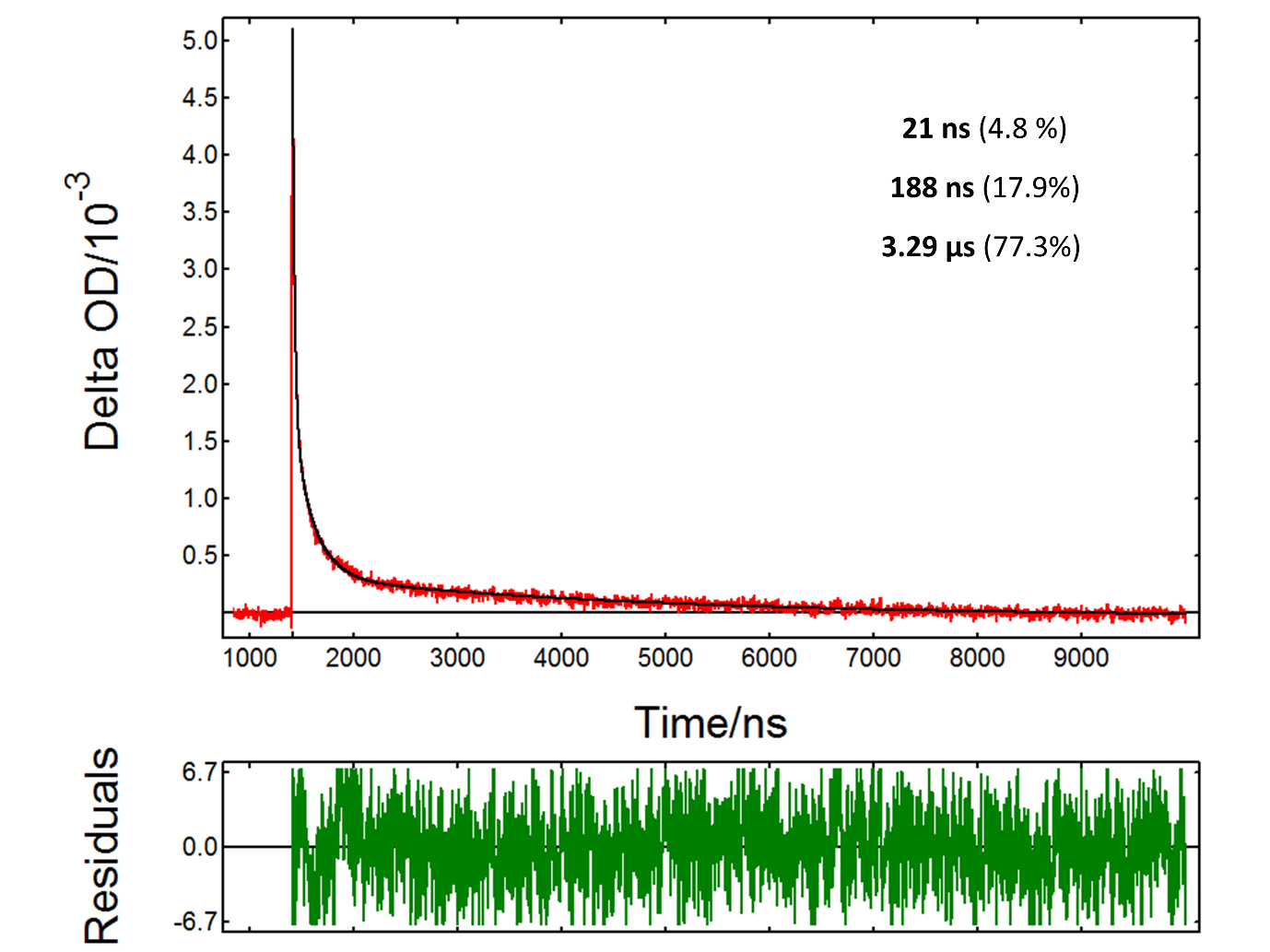
Figure 4: Transient absorption decay from a TiO2 suspension acquired in an LP980 spectrometer with a PMT detector: experimental data (red), fit result (black). Measurement conditions: λpump = 355 nm, λprobe = 390 nm, monochromator bandwidth = 0.50 nm, 5000 averages. The fit result is indicated in the graph.
If a suitable hole acceptor is added to an aqueous suspension of TiO2, it is possible to follow its reaction kinetics by transient absorption. In this application note we have chosen chloride anions, which react with surface trapped holes according to the following scheme5:
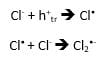
Cl– anions are provided by HCl in the solution. Their radical anion product Cl2•- has a transient absorption band in the UV extending into the visible.5 Figure 5 shows spectral TA data from a TiO2/HCl solution recorded under the same conditions as in Figure 3. In this case the spectrum is dominated by UV absorption with no TA present above 650 nm: the absorption is due to Cl2•- as opposed to trapped holes in TiO2. In order to test whether the hole transfer reaction above is taking place, a solution containing only HCl was measured and it did not show any transient absorption.
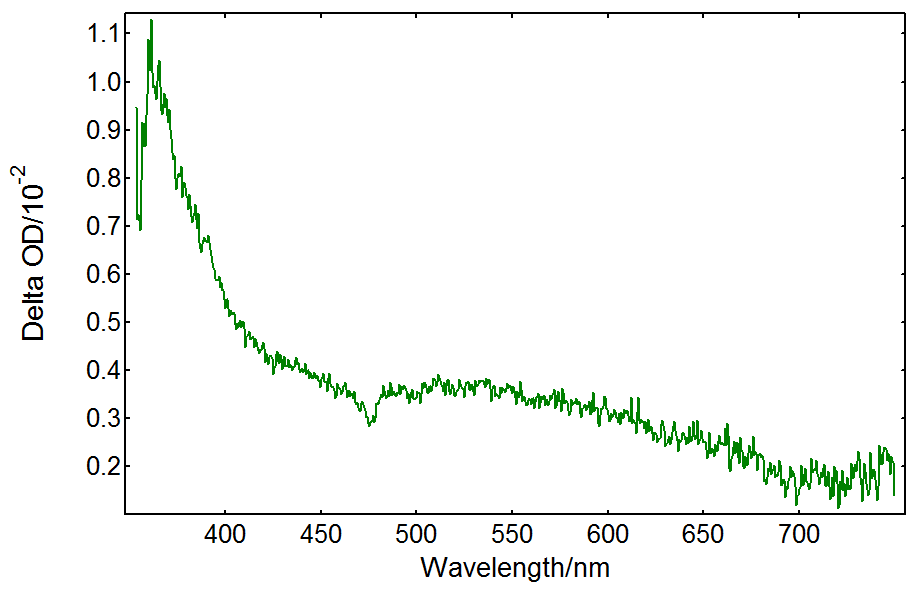
Figure 5: Transient absorption spectrum from a TiO2 suspension with 0.5 N HCl acquired in an LP980 spectrometer with an ICCD detector. Measurement conditions: λpump = 355 nm, monochromator bandwidth = 0.50 nm, ICCD delay = 100 ns, integration time = 1 µs, 50 averages.
The kinetic decay at 390 nm with HCl (Figure 6) is very different from the TiO2-only decay in Figure 4. In this case the absorption is dominated by the Cl2•- intermediate which decays over a longer timescale. The fact that TA occurs immediately after the flash suggests that Cl2•- is generated directly from Cl– adsorbed on the surface of TiO2. The decay is attributed to the reaction:![]()
Since it is a bimolecular process, it may be fitted to a second order kinetics model as follows:
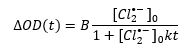
In the above equation [Cl2•-]0 is the initial concentration of chlorine radical anions, B is an experimental parameter and k is the reaction rate constant. The data were fit in FLASH, Edinburgh Instruments advanced analysis software for transient absorption.6 An initial concentration of 1.5 × 10-5 M was used based on previous studies of Cl2•- formation.5 A rate constant k = 9.8 × 108 M-1s-1 was obtained, which is in agreement with previous reports.7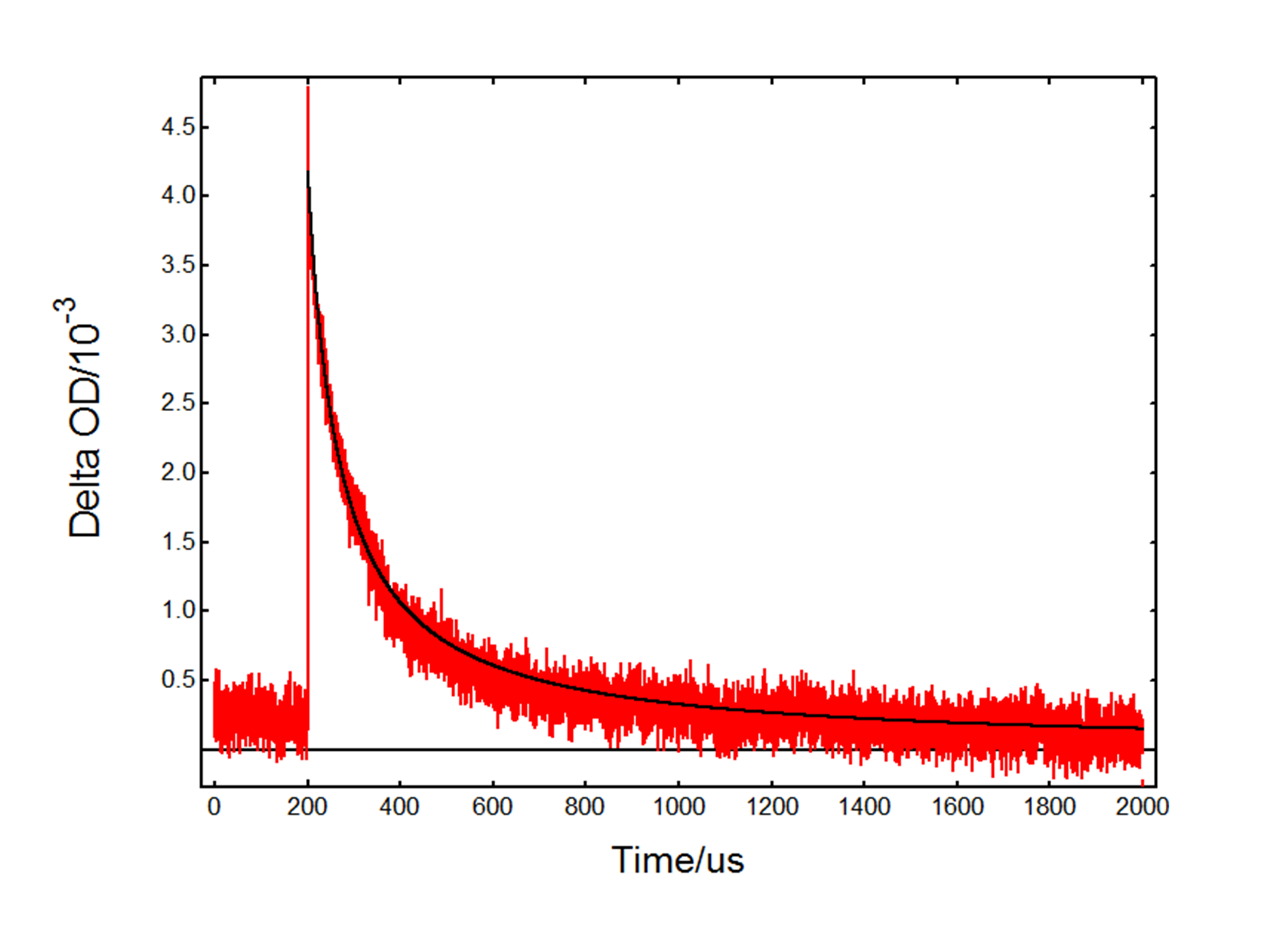
Figure 6: Kinetic transient absorption decay from a TiO2 suspension with added HCl, experimental data (red) and fit result (black). Measurement conditions: λpump = 355 nm, λprobe = 390 nm, monochromator bandwidth = 0.90 nm, 1600 averages.
Conclusions
The spectrum and kinetics of charge carriers in photoactivated TiO2, as well as the kinetics of a photocatalysed radical reaction, were studied in an LP980 Transient Absorption Spectrometer. Detailed spectral and time-resolved information was obtained thanks to the two detectors present in the LP980, a PMT and an ICCD camera. The results show that photogenerated holes in TiO2 decay in the microsecond scale with complex kinetics. Charge transfer between chloride anions and TiO2 initiates a radical reaction that takes place in the millisecond scale, as shown by transient absorption of the dichloride radical intermediate.
References
1. A. Fujishima, K. Honda, Nature 238 37-38 (1972)
2. J. Schneider, et al, Chem. Rev. 114 9919-9989 (2014)
3. S. Kohtani, A. Kawashima and H. Miyabe, Catalysts 7 303 (2017)
4. A. O. T. Patrocinio, et al, RSC Adv. 5 70536-70545 (2015)
5. J. Moser and M. Grätzel, Helvetica Chimica Acta 65 1436-1444 (1982)
6. FLASH, Edinburgh Instruments Ltd. (2012)
7. X. Yu, Z. Bao, and J. R. Barker, J. Phys. Chem. A 108 295-308 (2004)
